Predict the secondary structure for a single sequence
-
1. In this step, you'll predict the secondary structure for a tRNA, Sprinzl database RA7680.
The wizard mode of the GUI will be used, and this will predict the lowest free energy structure, the maximum expected accuracy structure, and the ProbKnot structure, which can contain pseuodoknots. All the structures will be color annotated by the base pairing probability.
-
2. Start the GUI.
-
3. Start the Input Window, Choose the Sequence, and Start the Calculation.
On the RNAstructure menu, choose "Structure Prediction->Single Sequence". This will open an input form.
Copy the example sequence from here to your local hard drive. This sequence is in fasta format. Help on the accepted input file formats is available in the online help (here).
In the "Predict a Secondary Structure Input Form", be sure that the button next to "RNA" is chosen next to "Sequence Type". (Alternatively, the secondary structure for a DNA sequence could be predicted by selecting "DNA".) Also, make sure sure the button next to "Load from File" is chosen next to "Sequence Source". Note that by choosing "Enter Sequence Below", a raw sequence can be pasted directly into the form. If a sequence is pasted, be aware that nucleotides in lowercase will be forced unpaired, therefore most or all nucleotides should be uppercase. Choose the input file by clicking the button "...". A file dialog will open that will let you choose RA7680.seq. This sequence choice will appear next to "Sequence File": and a default location for the output files will be chosen and will appear next to "Output File". When this is done, the input form will look like this
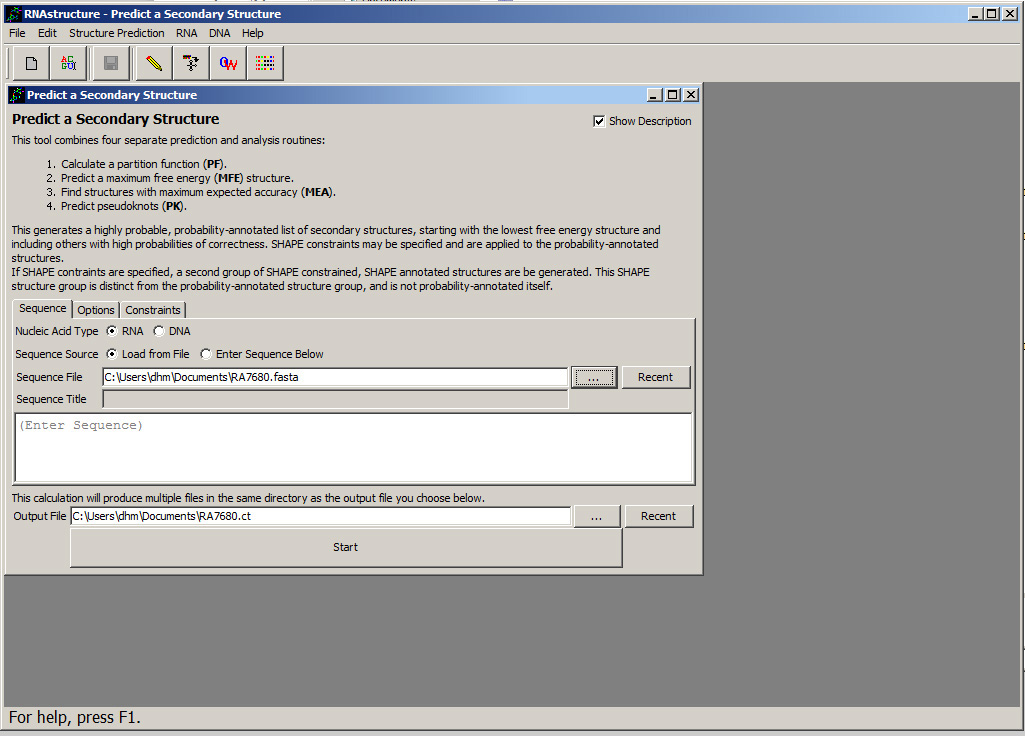 .
.At this time, the structure prediction options can be changed. Constraints or Restraints can be specified by clicking the "Constraints" tab. This are discussed in detail in the workshop step on improving secondary structure prediction accuracy using additional data. Also, the options for each program can be changed by clicking the "Options" tab. For this workshop, there is no need to change the options.
Start the calculation by clicking the "Start" button.
-
3. Viewing the Results.
Once the set of calculations are complete, the output form will appear, as shown here
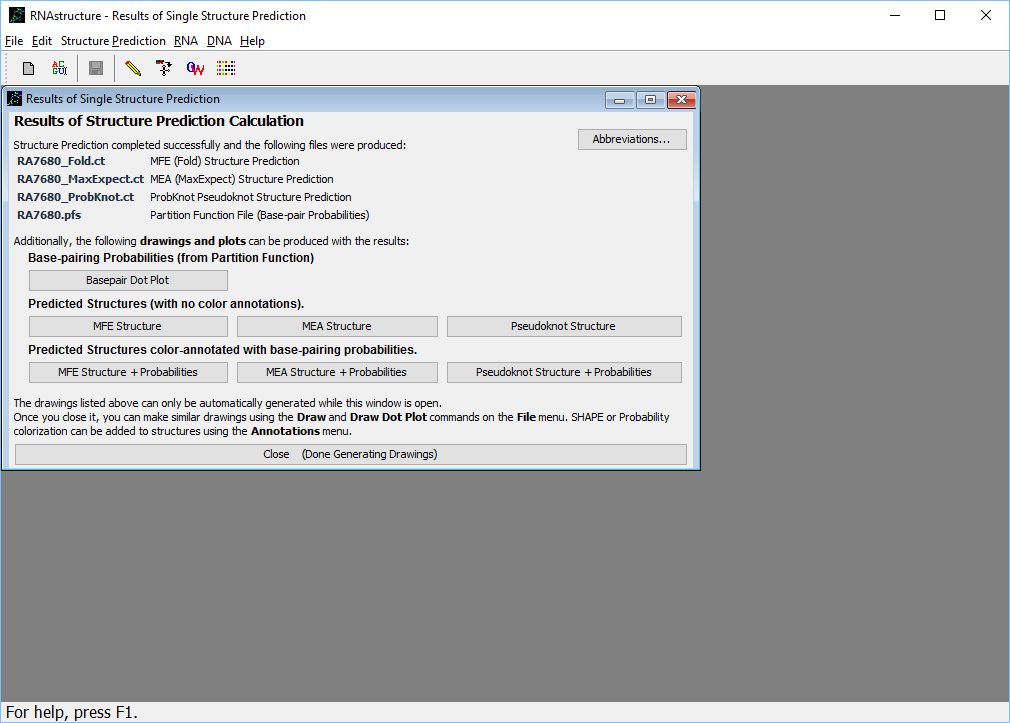 . Three different structure prediction methods were run, and the results of all three are available from this form. The MFE (minimum free energy) structure is the structure with lowest free energy and the most probable structure. The MEA (maximum expected accuracy) structure is composed of the most probable base pairs. The "Pseudoknot Structure" is generate by a program called ProbKnot, and it can contain pseudoknots (but MFE and MEA cannot contain pseudoknots). The three methods might predict different structures, and these three results represent alternative hypotheses for the structure. The base pairs predicted in the MEA structure, on average, are slightly more likely to be correct than those predicted in the MFE structure. The "Pseudoknot structure" can be consulted to see if there are possible pseudoknots.
. Three different structure prediction methods were run, and the results of all three are available from this form. The MFE (minimum free energy) structure is the structure with lowest free energy and the most probable structure. The MEA (maximum expected accuracy) structure is composed of the most probable base pairs. The "Pseudoknot Structure" is generate by a program called ProbKnot, and it can contain pseudoknots (but MFE and MEA cannot contain pseudoknots). The three methods might predict different structures, and these three results represent alternative hypotheses for the structure. The base pairs predicted in the MEA structure, on average, are slightly more likely to be correct than those predicted in the MFE structure. The "Pseudoknot structure" can be consulted to see if there are possible pseudoknots.For this sequence, all three programs predict exactly the same structure. Click on the button labeled "MFE Structure + Probabilities". This opens the structure viewing window, as shown here
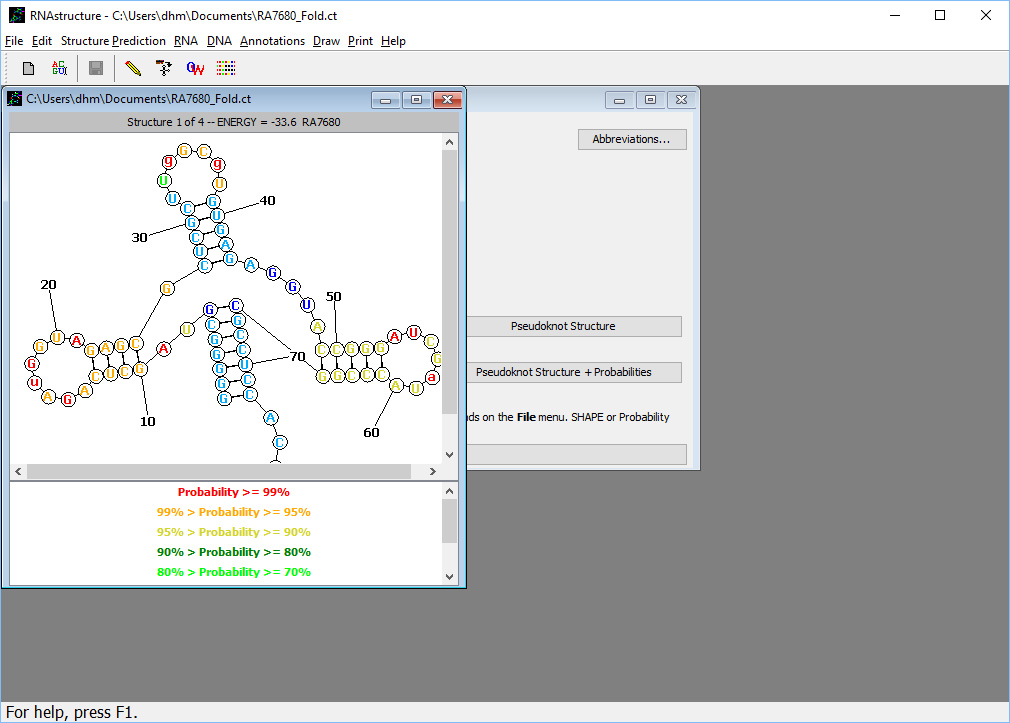 . The predicted secondary structure is shown where nucleotides appear in circles, and lines are used to show the path of the backbone and base pairs. Each nucleotide is color annotated by probability. Nucleotides that are base paired are colored according to the estimated probability of the base pair. Unpaired nucleotides are colored according to the estimated probability of being unpaired. Base pairs of high probability are more likely to be correctly predicted base pairs. For example, about 2/3 of predicted base pairs, on average, are correct predictions. But 90% of the base pairs of 99% or higher pairing probability, on average, are correct predictions.
. The predicted secondary structure is shown where nucleotides appear in circles, and lines are used to show the path of the backbone and base pairs. Each nucleotide is color annotated by probability. Nucleotides that are base paired are colored according to the estimated probability of the base pair. Unpaired nucleotides are colored according to the estimated probability of being unpaired. Base pairs of high probability are more likely to be correctly predicted base pairs. For example, about 2/3 of predicted base pairs, on average, are correct predictions. But 90% of the base pairs of 99% or higher pairing probability, on average, are correct predictions.The MFE structure prediction also predicted suboptimal structures, which are low free energy structures. These are important as alternative hypotheses for the structure. To view suboptimal structures, press "control-up arrow" ("control-command-up arrow" on Mac OS X). Structure 2 of 4 with folding free energy change of -32.5 kcal/mol will be shown. "Control-up arrow" ("control-command-up arrow" on Mac OS X) increases the structure index shown and "control-down arrow" ("control-command-down arrow" on Mac OS X)decreases the index, where structures are sorted by folding free energy change.
Under the "Draw" menu iitem, there are a number of options. "Go to Structure..." allows choice of the index of the displayed structure. "Zoom" controls the size of the drawing. Pressing "control-left arrow" and "control-right arrow" zooms out and in, respectively. "Render Circular Structure" changes the structure drawing to an alternative form where the backbone is around a circle, and pairs are indicated by lines. By default, a structure with a pseudoknot is displayed this way. "Write Dot-Bracket file" can be used to write the structure in dot-bracket format. By default, RNAstructure saves structures in .ct format. "Write Postscript File" and "Write SVG File" can be used to write vector drawings that can be edited by programs such as Illustrator or Inkscape.
Back to Workshop Home or Continue to Next Step
On Windows, use the start menu and choose "all programs->RNAstructure 6.0->RNAstructure".
On Mac, double-click the RNAstructure app.
On Linux, execute RNAstructureScript (in the exe/ subdirectory).