Improve the Accuracy of Secondary Structure Prediction Using Experimental Data
-
1. In this step, you'll predict the secondary structure for the group I intron P546 domain with and without experimental information. The experimentaol data improves the structure prediction accuracy.
SHAPE is an experimetal method that can provide information about whether nucleotides are paired or unpaired. Unpaired nucleotides tend to be more reactive to SHAPE.
Copy the following files to your local hard drive: the P546 sequence in FASTA format and the P546 SHAPE mapping data, collected by the Weeks lab.
-
2. Use the GUI to predict the secondary structure of P546.
-
3. Use the GUI to predict the secondary structure of P546, using SHAPE restraints.
On the RNAstructure menu, choose "Structure Prediction->Single Sequence". This will open an input form.
Select the sequence by clicking the "..." button next to "Sequence File".
Now, click the tab labeled "Constraints". This form (shown here
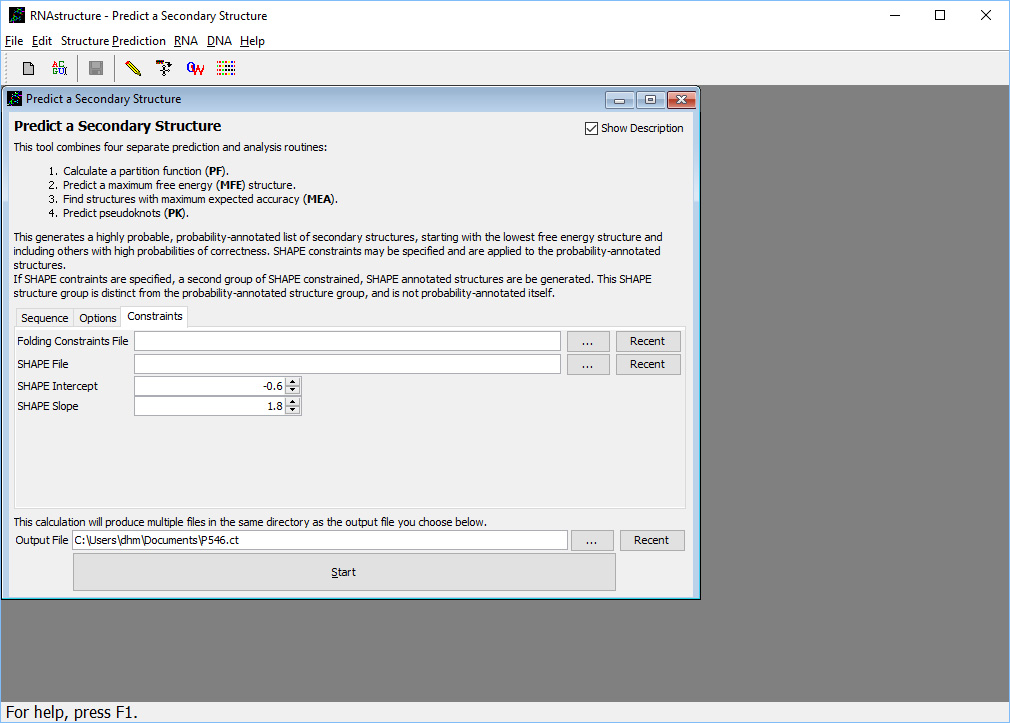 ) allows the input of several types of constraints. A folding constraints file can specify nucleotides that must be base paired, nucleotides that muct be unpaired, base pairs that must occur, and base pairs that are not allowed. In this workshop, we will not be using folding constraints, but will instead improve structure prediction using SHAPE restraints.
) allows the input of several types of constraints. A folding constraints file can specify nucleotides that must be base paired, nucleotides that muct be unpaired, base pairs that must occur, and base pairs that are not allowed. In this workshop, we will not be using folding constraints, but will instead improve structure prediction using SHAPE restraints.Enter the SHAPE file name to use the SHAPE data by clicking the "..." button next to "SHAPE File". Use the P546 SHAPE file you copied in step 1. The file format is specified here. Two parameters are used to control the effect of the SHAPE data on the structure prediction, SHAPE intercept and SHAPE Slope. These parameters were found by optimizing accuracy using SHAPE data for set of seqeunces with known structure, and the default values should work well for most calculations.
Start the calculation by clicking the "Start" button.
-
3. Viewing the Results.
From the output form, click the button labeled "MFE Structure+Probabilities". This predicted structure is now highly accurate as compared to the known structure. Also, the quality of the predicted pairs is higher in that almost all predicted base pairs are estimated to have pairing probability of 80% or higher. The CircleCompare plot comparing the prediction with SHAPE to the known structure is available here.
Back to Workshop Home or Continue to Next Step
s
First, predict the structure without using restraints. Follow the procedure for predicting a secondary structure from step 2 of the workshop. Use "Structure Prediction->Single Sequence".
P546 is an example sequence that has a relatively poorly predicted secondary structure without using additional information. You can compare the predicted structure to the accepted structure, P546-correct.ct. The accepted structure can be drawn in the RNAstructure GUI by choosing on the menu and then choosing this ct file. Note that the pairs in the MFE prediction that are incorrect are all estimated to have low (<50%) pairing probability. A convenient way to compare structures is to use the RNAstructure CircleCompare program (for reference, this program runs on the command line or the web server, but the results are provided for the workshop). This shows an accepted and predicted structure on the same circular drawing, where pairs in green are in both structures, pairs in red are in the predicted structure only, and pairs in black are in the accepted structure only. The circle compare drawing for the MFE prediction of P546 compared to the known structure is available here.