|
orega evolves an input sequence to increase end-to-end distance by avoiding base pairing using a genetic algorithm. Only nucleotides in a specified segment are mutated.
USAGE: orega <input file> <start> <length> <output
file> [options]
| <input file> |
The name of a sequence
file containing input data.
Note that lowercase nucleotides are forced single-stranded
in structure prediction. |
| <start> |
Integer that indicates the first nucleotide that can be mutated. |
| <length> |
Integer that indicates the length of the sequence segment that canb be mutated. |
| <output file> |
The name of a FASTA file to which the output will be written. |
--nocomplexity
|
Use the objective function that does not include sequence complexity. The default uses an objective function that includes sequence complexity. (This is maintained for backwards compatability. In practice, -f should be used to select the objective function.) |
-d, --dna
|
Use DNA paarmeters for folding. The default is RNA parameters. |
| --filterAUG |
Sequences that have a premature AUG start codon will be eliminated from the population. The default is to not test for AUG start codons. |
| --filterCUG |
Sequences that have a premature CUG start codon will be eliminated from the population. The default is to not test for CUG start codons. |
| -h --help |
Display the usage details message.
|
| --limitG |
Nucleotide G will not be an option during nucleotide mutation. The default is to allow mutation to G.
In practice, --limitG accelerates the search. |
| --MutationSwitch |
Mutation site will be based on the mean base pair probability at specific nucleotide instead of random selection. This bias change towards nucleotides of higher base pairing probability. The default is to choose any nucleotide at random for mutation.
In practice, --MutationSwitch accelerates the search for sequences. |
-v --version
|
Display version and copyright information for this
interface. |
| -a, -A, --alphabet |
Specify the name of a folding alphabet and associated
nearest neighbor parameters. The alphabet is the prefix
for the thermodynamic parameter files, e.g. "rna" for RNA
parameters or "dna" for DNA parameters or a custom
extended/modified alphabet. The thermodynamic parameters
need to reside in the at the location indicated by
environment variable DATAPATH.
The default is "rna" (i.e. use RNA parameters).
A list of available folding alphabets is here. |
| --ComplexityConstant |
The A value in the objective function (see below) that weights the linguistic complexity. Default is 1.0. |
| -f, --func |
The objective function, an integer that chooses which objective function to use. 1 = SIMPLE (no sequence complexity used), 2 = COMPLEX (the sequence complexity is used). 3 = ALL (additional sequence pair is used). The default is 2. |
| --filteroligoA |
This options specified the maximum number of As in a row. If this is exceeded in a sequence, that sequence will be eliminated from the population. The default value is 0, which indicates no filtering will occur. |
-i, --iter
|
The number of iterations (optimization steps) to run. The default is 1000.
|
-mr, --mutate
|
The mutation rate, the probability that a nucleotide in the target segment should be mutated. The default is 0.03. |
| -n, --population |
The population size, the number of concurrent sequences used in the genetic algorithm. The default is 10. |
| -rf, --recomb |
The recombination frequency, the number of iterations that are run before a recombination/crossover step occurs. The default is 6. |
| --restart |
Specify the name of a previous state file (created with the --save option) that should be loaded to restart an optimization from where it left off before, or continue optimizing a previous result. |
| -rr, --crossover |
The recombination rate, the probability that a nucleotide will be selected as a recombination marker. The default is 0.03. |
| -rs, --seed |
Specify a random seed. This is required to get exactly reproducible results. The default is to use a seed based on the current system time. |
| -sav, --save |
Specify the name of a file where intermediate results can be saved. This file can be used to restart the calculation. |
| -t |
The threshold to determine if a nucleotide is considered base paired or not. This function will be only used when objection function -f = 3. Default is 0.4 |
orega indirectly increases the end-to-end distance of a sequence by evolving a sequence segment to avoid base pairs. The objective function by default also includes the sequence complexity, and this is helpful to keep the sequence from evolving into repeats.
The default objective function (-f 2) is:
Fitness score = (1 – avgP) + A×c
where avgP is the average base pairing probability in the segment, A is a weight for sequence complexity (set by --ComplexityConstant, default 1.0), and c is the linguistic sequence complexity.
The full objective function (-f 3) is:
Fitness score = (1 – avgP) + A×c + (N - sum of base pairs)
where N is the number of nucleotides in the fragment and sum of base pairs is the number of base pairs for nucleotides in the fragment with a probability above the threshold (set by -t with default of 0.4).
The lean objective function (-f 1) is:
Fitness score = (1 – avgP)
Linguistic complexity is calculated using (Gabrielian & Bolshoy, 1999):
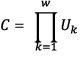
where Uk is the fraction of possible sequences observed for a k-mer and w is the maximum k-mer size. Uk is:
Uk=min[4k,L-k+1]
where L is the sequence length. w is adopted based on sequence length with w=3 for L<18, w=4 for 18≤L<67, w=5 for 67≤L<260, w=6 for 260≤L<1029, and w=7 for L≥1029.
The complexity calculation performed by orega assumes a 4-nucleotide alphabet (although RNAstructure can use larger alphabets to include modified nucleotides). It is important to provide a sequence that uses only the standard A, C, G, U/T nucleotide alphabet. Lowercase nucleotides are forced unpaired (as in the rest of RNAstructure), but these should only be included outside the region of sequence being evolved. Lowercase nucleotides within the evolved region will never be allowed to pair as the sequece evolves.
orega-cuda is the cuda version (for execution on cuda-enabled graphics cards). The same options are used, but the cuda-enabled partition function is used in the background. This can dramatically improve runtimes.
- Lai, W. C., Kayedkhordeh, M., Cornell, E. V., Farah, E., Bellaousov, S., Rietmeijer, R., Mathews, D. H., & Ermolenko, D. N.
mRNAs and lncRNAs intrinsically form secondary structures with short end-to-end distances.
Nature Communications, 9: 4328. (2018).
- Gabrielian, A. & Bolshoy, A.
Sequence complexity and DNA curvature.
Computational Chemistry, 2: 263-74. (1999).
- Reuter, J.S. and Mathews, D.H.
"RNAstructure: software for RNA secondary structure prediction
and analysis."
BMC Bioinformatics, 11:129. (2010).
|