OligoWalk Instructions
II. Input Options
III. Output
IV. Calculation Time
V. References
I. Introduction
The OligoWalk web server is constructured to predict efficient siRNA candidates for an mRNA sequence on the basis of the statistical mechanics of the siRNA-target interaction. The secondary structures of the oligomer and target mRNA are considered in the OligoWalk algorithm. The euquilibrium is:
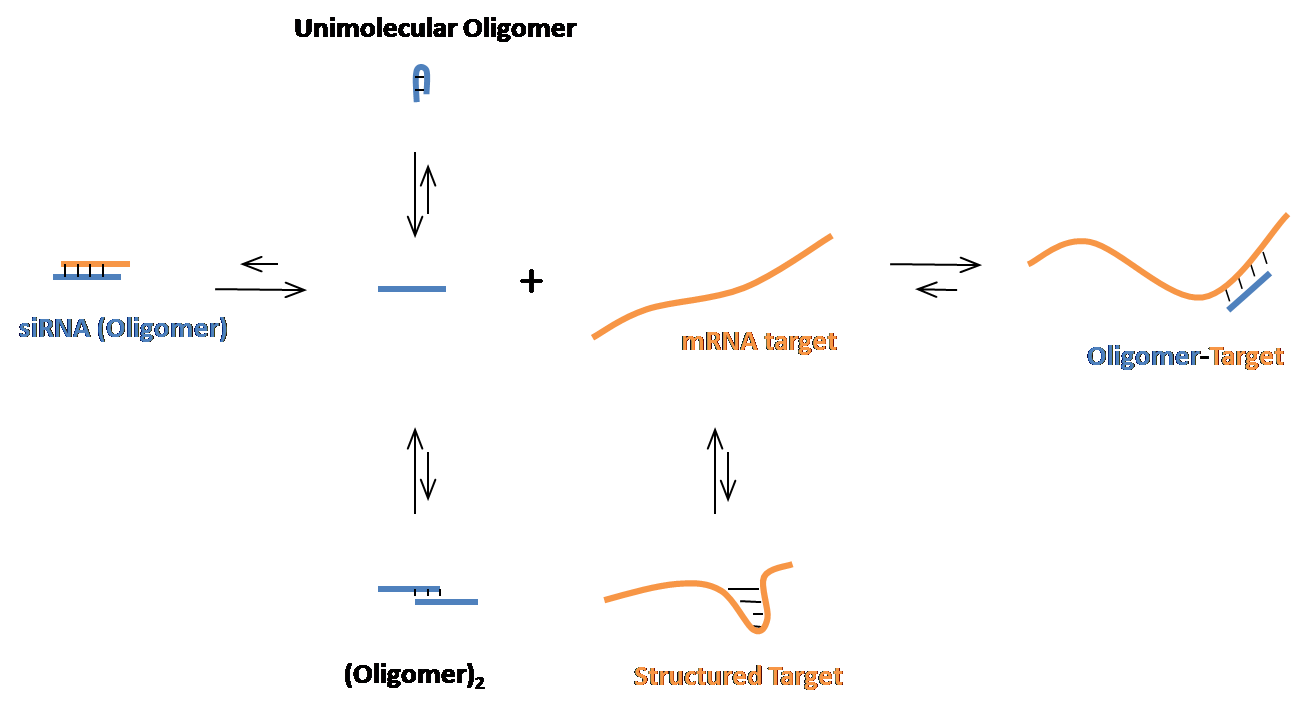
In addition to the thermodynamic parameters, a number of local sequence features are also considered by the Support Vector Machine embedded in the server. Please see References for the details.
The input options are restricted to save the calculation time on server. The complete software package without restrictions is available for download.
Oligomer Type: Only RNA is allowed.
Oligomer Length: 19 bases is recommended for siRNA design.
Oligomer Concentration: This is the oligomer concentration. It only affects overall ΔG that is provided as part of the thermodynamic details and it is not used for siRNA selection.
Prefilter for siRNA design: The prefiter is modified from Reynold's method (see References). The prefilter only allows candidate siRNAs that meet some basic sequence criteria to be considered using the full thermodynamic model. It must be turned on when the secondary structure of the target is considered in order to save time.
Binding mode: Three modes are available.
"Break local structure": The target will be folded once. The base pairs of the binding region are broken for the hybridization of the oligomer, but the structures of the other parts will not be refolded. This mode is a compromise between the fastest and most rigorous methods.
"Refold target RNA" : A certain range of region (the size is defined by Folding size, specified below) centered on the binding site will be refolded after oligomer hybridization. This mode is the most rigorous if it is assumed that the system is at equilibrium and this is the recommended mode for siRNA selection.
"Not consider target structure": not consider the secondary structure of the target. This is the fastest mode.
Folding option: Three options are available.
"Only consider the Optimal Structure": Only the lowest free energy structure is considered for the secondary structure prediction of RNA.
"Consider Suboptimal Structures": Suboptimal structures are also considered using a heuristic method to find suboptimal RNA secondary structures.
"Consider all possible structures using partition funcion": All possible structures are considered with a partition function calculation. This method is the most rigorous and is recommended for siRNA selection.
Folding size: When secondary structure of the taget is selected to be considered ("Break local structure" or "Refold target RNA"), the user can define the folding size of target. Then, a certain number of nucleotides centered at the binding region are considered for the secondary structure prediction.
Scan region: The positions on the target region to be scanned to find the candidates. By defining a sub-region of the sequence, considerable compute time can be saved..
III. Output
The server generates an siRNA candidate table ranked by the probability of being efficient at knock-down. It also outputs a thermodynamic table:
1. Overall ΔG: The net free energy change when all contributions in the above figure are considered. The target-siRNA complex product prefers negative Overall ΔG .
2. Duplex ΔG : The free energy change of hybidized duplex between oligomer and target. Stable duplex has more negative ΔG than unstable one.
3. Tm-dup: The melting temperature in degrees C for the hybrided duplex formation,
4. Break-targ. ΔG: The free energy cost for opening base pairs in the region of complementarity to the target. More negative value means it is less accessible for siRNA binding.
5. Intraoligo ΔG: The free energy change of the self structure of unimolecular oligo. Stable self structure has more negative ΔG than unstable one
6. Interoligo ΔG: The free energy change of oligo-oligo dimer. Stable dimerization has more negative ΔG than unstable one
7. Number of structures: The number of suboptimal structures of target used before and after the binding of oligomer. This is only available when "Consider Suboptimal Structures" is selected.
8. End-diff. : The free energy difference between the 5' and 3' end of the antisense strand of siRNA, with windows of 2. It has been shown that a functional siRNA prefer to have an unstable 5' end (End-diff is more positive).
9. Prefilter score: The score calculated using Reynold et al. method.
IV. Calculation Time
Calculation time and memory size :
Target mRNA (Genbank ID) |
Sequence length (nucleotide) |
Time a |
Memory |
NM_020548 |
730 b |
0:57:17 |
93 |
M60857 |
851 |
3:55:53 |
110 |
NM_002870 |
1211 |
6:09:25 |
112 |
NM_002467 |
2189 |
6:36:42 |
113 |
AJ272212 |
3460 |
6:53:05 |
117 |
The benchmarks were performed with the default options: The oligonucleotide was a 19 base RNA; the folding size of the target was 800 nucleotides centering on the binding site (full length if the whole target has less than 800 nt); the partition function calculation was conducted; the entire mRNA was scanned; the prefilter was turned on. The time cost is similar for long sequence because the prefitler (7) is turned on and number of candidates being folded is limited. |
|||
Binding Mode |
Folding Option |
Time (h:min:sec) |
Memory (MB) |
Not Consider Target Structure |
NA |
<0:00:01 (0:00:04) |
<1 |
Break Local structure |
Optimal Structure |
0:00:53 (0:00:54) |
15 |
Suboptimal Structure |
0:01:40 (0:05:11) |
15 |
|
Refold target RNA |
Optimal Structure |
0:40:09 (3:48:42) |
15 |
Suboptimal Structure |
1:51:56 (10:02:55) |
15 |
|
Partition Function |
0:57:17 (3:43:26) |
93 |
|
The caculation is tested on mRNA NM_020548 which is 730 nucleotides in length. The oligomer type is RNA. The oligomer length is 19 bases. The folding size is all, the full length of the mRNA. The scanned region size is from the first base to the end. There are 712 and 132 oligomer candidates generated without and with prefilter turned on, respectively. The calculation time without prefilter is shown in parenthesis. |
|||
V. References
The OligoWalk algorithm is based on the following research:
The efficient siRNA selection method is described in :
Z.J. Lu and D.H. Mathews. (2008) "Efficient siRNA selection using hybridization thermodynamics"
Nucleic Acids Research, 36:640-647.
The siRNA prefilter method is described in :
Reynolds, A., Leake, D., Boese, Q., Scaringe, S., Marshall, W.S. and Khvorova, A. (2004) Rational siRNA design for RNA interference. Nat Biotechnol., 22, 326-330.
The thermodynamic parameters and algorithm of RNA secondary structure prediction are described in:
Lu, Z.J., Turner, D.H. and Mathews, D.H. (2006) A set of nearest neighbor parameters for predicting the enthalpy change of RNA secondary structure formation. Nucleic Acids Res., 34, 4912-4924.
D.H. Mathews, M.D. Disney, J.L. Childs, S.J. Schroeder, M. Zuker, and D.H. Turner. "Incorporating Chemical Modification Constraints into a Dynamic Programming Algorithm for Prediction of RNA Secondary Structure"
Proceedings of the National Academy of Sciences USA, 101, 7287-7292, (2004).
D.H. Mathews, J. Sabina, M. Zuker, and D. H. Turner "Expanded Sequence Dependence of Thermodynamic Parameters: Improves Prediction of RNA Secondary Structure" Journal of Molecular Biology, 288, 911-940, (1999).
The thermodynamic parameters of DNA/RNA duplex :
N. Sugimoto, S. Nakano, A. Katoh, H. Nakamura, T. Ohmichi, M. Yoneyama, and M. Sasaki
"Thermodynamic Parameters to Predict the Stability of RNA/DNA Hybrid Duplexes"
Biochemistry, 34, 11211-11216, (1995).
Other research related to OligoWalk:
Ladunga I. "More complete gene silencing by fewer siRNAs: transparent optimized design and biophysical signature" Nucleic Acids Res 35:433-440, (2007)
O.V. Matveeva, D.H. Mathews, A.D. Tsodikov, S.A. Shabalina, R.F. Gesteland, J.F. Atkins, S.M. Freier
"Thermodynamic criteria for high hit rate antisense oligonucleotide design"
Nucleic Acids Res., 31, 4989-4994, (2003).
D.H. Mathews, M.E. Burkard, S.M. Freier, J.R. Wyatt, and D.H. Turner
"Predicting Oligonucleotide Affinity to Nucleic Acid Targets"
RNA, 5, 1458-1469 (1999).
OligoWalk was trained and tested on two experimental databases:
Huesken D, Lange J, Mickanin C, Weiler J, Asselbergs F, Warner J, Meloon B, Engel S, Rosenberg A, Cohen D, Labow M, Reinhardt M, Natt F, Hall J. (2005). Design of a genome-wide siRNA library using an artificial neural network. Nat Biotechnol 23:995-1001.
Shabalina SA, Spiridonov AN, Ogurtsov AY. (2006). Computational models with thermodynamic and composition features improve siRNA design. BMC Bioinformatics 7:65.